
Die anodische Auflösung von Aluminium in Lewis-sauren ionischen Flüssigkeiten, bestehend aus Aluminiumchlorid und 1-Ethyl-3-methylimidazoliumchlorid, wurde mittels linearer Polarisation und zyklischer Voltammetrie, der elektrochemischen Quarzkristall-Mikrowaage (EQCM) und Chronopotentiometrie bei Umgebungstemperatur untersucht. In einem 2:1-Elektrolyten wurde eine anodische Passivierung der Arbeitselektrode beobachtet, während in einem 1,5:1-Elektrolyten keine Passivierung auftrat. Die Chronopotentiometrie belegt, dass die Passivierung durch eine lokale Verfestigung des Elektrolyten aufgrund einer Erhöhung der Aluminiumkonzentration in der Nähe der Anode verursacht wird. EQCM-Daten unterstützen diese Ergebnisse.
Anodic dissolution of aluminum and anodic passivation in [EMIm]Cl-based ionic liquids
The anodic dissolution of aluminum in Lewis acidic ionic liquids consisting of AlCl3 and 1-ethyl-3-methylimi-dazolium chloride was studied using linear sweep and cyclic voltammetry, an electrochemical quartz crystal microbalance (EQCM) and chronopotentiometry at ambient temperature. Anodic passivation of the working electrode was observed in a 2:1 electrolyte while no passivation was found in a 1.5:1 electrolyte. Chronopotentiometry proves the passivation to be caused by local solidification of the electrolyte due to an increase in the aluminum concentration near the anode. EQCM data support these results.
1 Einleitung
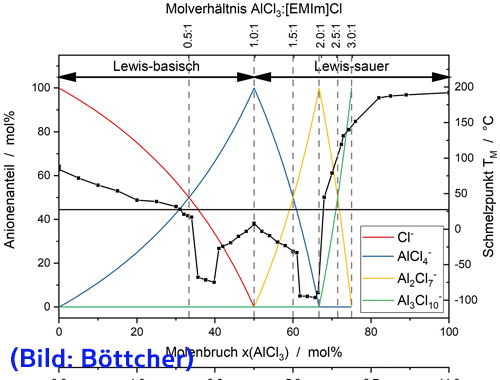
Elektrochemisch abgeschiedenes Aluminium beziehungsweise Aluminiumlegierungen stellen sehr interessante Beschichtungen dar. Vor allem die Abscheidung aus ionischen Flüssigkeiten hat sich als erfolgversprechend herausgestellt [1]. Mischungen aus Aluminiumchlorid (AlCl3) mit Salzen auf Basis von Imidaozolium, beispielsweise 1-Ethyl-3- methylimidazolchlorid ([EMIm]Cl), wurden intensiv untersucht. Die Elektrolyte zeigen eine gute Löslichkeit für unterschiedliche Metallsalze, was die Abscheidung von Aluminium und zahlreichen Aluminiumlegierungen erlaubt [2–5]. Die Lewis-Azidität der Schmelze kann über eine Änderung des Molverhältnisses von [EMIm]Cl und AlCl3 eingestellt werden, wodurch die vorherrschenden Anionen in der Schmelze bestimmt werden (Abb. 1). Es muss eine Lewis-saure Schmelze vorliegen, um die Abscheidung von Aluminium gemäß Gleichung <1> zu ermöglichen [1-6, 8]:
4Al2Cl7- + 3e- → Al + 7AlCl4-<1>
Zur Erzielung eines kontinuierlichen Abscheidevorgangs sowie zur Vermeidung der anodischen Zersetzung der ionischen Flüssigkeit können lösliche Aluminiumanoden eingesetzt werden. Allerdings nimmt die Abscheidespannung bei der Verwendung von löslichen Aluminiumanoden während der galvanostatischen Abscheidung aus einem 2:1-Elektrolyten bei Raumtemperatur zu, wenn eine kritische anodische Stromdichte von 8,5 mA/cm2 überschritten wird. Der Abscheideprozess wird unterbrochen, sobald die Maximalspannung der Spannungsversorgung erreicht ist. Dieser Effekt kann umgangen werden, wenn Anoden mit großer aktiver Fläche verwendet werden oder der Elektrolyt stark gerührt wird [9]. Aufgrund der ungleichmäßigen Verteilung des elektrischen Felds auf der Anode können trotzdem lokale Bereiche vorliegen, bei denen der kritische Wert der Stromdichte überschritten wird. Dies führt ebenfalls zum Anstieg der Abscheidespannung.

Abb. 1: Theoretische ionische Zusammensetzung (farbige Kurven) und Phasendiagramm (schwarze Linie mit ausgefüllten Quadraten) von [EMIm]Cl und AlCl3 [22–24]; die vertikalen, gestrichelten Linien stellen die Elektrolytzusammensetzung dar, ausgedrückt als Molverhältnisse von AlCl3 zu [EMIm]Cl, und die horizontale, durchgezogene Linie markiert den Wert für die Umgebungstemperatur in der Handschuhbox (die Interpretation der Farbverweise in der Abbildungslegende kann der Webversion dieses Artikels entnommen werden)
Die vorliegende Arbeit konzentriert sich auf die Auflösung von Aluminium in Lewis-sauren ionischen Flüssigkeiten aus [EMIm]Cl/AlCl3 mit dem Ziel, den oben angesprochenen Mechanismus der Anodenpassivierung besser zu verstehen. Potentiodynamische Polarisationsuntersuchungen sowie zyklische Voltammetriemessungen wurden durchgeführt, um den Grund für die Begrenzung der anodischen Stromdichte bei der Abscheidung aufzuklären. Die Kombination aus elektrochemischen Quarzkristall-Mikrowaage (EQCM) und der Anwendung der Theorie von Sand zum Konzentrationsprofil an der Elektrode bei konstanter Stromdichte [10] wurden herangezogen, um den Anstieg der Zellspannung bei hohen anodischen Stromdichten bei den verwendeten löslichen Aluminiumanoden zu erklären.
Galvanostatische Sprungexperimente sind leistungsstarke Methoden zur Untersuchung der Eigenschaften der Metallabscheidung und Metallauflösung. Abhängig von der Höhe des Stromsprungs liefert der resultierende Transient wertvolle Informationen über den Diffusionskoeffizienten der elektrochemisch aktiven Spezies [10, 11], das Keimbildungsverhalten [6, 12] und den Ladungstransfer [13].
Die EQCM ist eine in-situ Methode, die es ermöglicht, elektrochemische Messungen mit einer genauen Bestimmung der abgeschiedenen Masse zu kombinieren. Die Abnahme der Resonanzfrequenz Df eines Quarzkristalls hängt mit der Zunahme der flächenbezogenen Massendichte m gemäß der Sauerbrey-Gleichung (Gl. <2>) zusammen [14, 15]:
 <2>
<2>
Berührt eine Seite des Resonators eine Flüssigkeit der Dichte r1 und Viskosität h1, so kommt es zu einer zusätzlichen Frequenzabnahme gemäß der Kanazawa-Gleichung (Gl. <3>) [14, 16]:
 <3>
<3>
In Gleichung <2> und <3> ist f0 die Resonanzfrequenz des unbeladenen Quarzes und rq und µq sind die Dichte beziehungsweise der Schermodul des Quarzes. Um Gleichung <2> anwenden zu können, muss die Dämpfungsänderung des Schwingquarzes Dw kleiner sein als die Änderung der Resonanzfrequenz Df. Im Ansatz der Autoren des vorliegenden Beitrags wird die Änderung der Dämpfung des Quarzkristalls als Änderung der Halbwertsbreite (full width half maximum, FWHM) des Quarzkristalls nahe seiner Resonanzfrequenz gemessen [17]. Nach Gleichung <3> bewirkt eine Zunahme der Viskosität der Flüssigkeit hl in Kontakt mit dem Quarzkristall eine Abnahme der Frequenz und eine Zunahme der Dämpfung.
2 Versuchsdurchführung
1-Ethyl-3-methylimidazoliumchlorid, [EMIm]Cl, (> 98 %, Iolitec, Deutschland) wurde über einen Zeitraum von zwei Tagen bei 60 °C getrocknet, um einen Feuchtigkeitsgehalt unter 100 ppm zu erreichen (bestimmt durch Karl-Fischer-Titration, Modell 831 KF-Coulometer, Metrohm, Deutschland [18]). Wasserfreies Aluminiumchlorid (AlCl3; Granulat, 99 %, abcr, Deutschland) wurde ohne weitere Reinigung verwendet. Die Elektrolyte wurden durch langsame Zugabe von Aluminiumchlorid zu [EMIm]Cl in Molverhältnissen von 0,5:1 bis 2,0:1 hergestellt, die im Folgenden als x:1-Elektrolyte bezeichnet werden (x = 0,5, 1,0, 1,5, 2,0). Das Rühren der Mischung über einen Zeitraum von 24 Stunden ergab je nach Zusammensetzung transparente bis leicht gelbe Flüssigkeiten.
Die elektrochemischen Untersuchungen wurden in einer mit Argon gefüllten Glovebox (VAC Atmospheres, USA, O2 < 0,5 ppm, H2O < 0,5 ppm) unter Verwendung eines SP300- oder VSP-Potentiostaten/Galvanostaten (BioLogic, Frankreich) durchgeführt. Als Arbeitselektrode (AE) wurde ein Aluminiumdraht (99,999 %, Alfa Aesar) von 1 mm Durchmesser verwendet, der in ein Glasröhrchen eingeschoben und mit Epoxidharz (Epoxy 2000 Plus, Cloeren Technology, Deutschland) fixiert wurde. Als ringförmige Gegenelektroden (GE) wurden Aluminiumbleche von 2 mm Dicke (99,0 %, Goodfellow) und als Referenzelektrode (RE) ein Aluminiumdraht (99,999 %, Alfa Aesar) verwendet. Im Folgenden sind alle Potenziale in Bezug auf diese Referenz angegeben. Vor jedem Experiment wurde die Arbeitselektrode mit Schleifpapier (SiC, 800–4000 Körnung) bearbeitet, um eine reproduzierbare Qualität der Elektrodenoberfläche zu erhalten.
Für die EQCM-Messungen wurden polierte Quarze im AT-Schliff mit einer Resonanzfrequenz von 10 MHz und Goldelektroden von etwa 100 nm Dicke (KVG, Deutschland) verwendet. Die Resonanzfrequenz und die Dämpfung des Quarzkristalls wurden mit einem Netzwerkanalysator (Agilent E5100A) gemessen, während für die elektrochemischen Messungen ein Potentiostat/Galvanostat Modell 263A (EG&G Princeton Applied Research) verwendet wurde [19].
3 Ergebnisse und Diskussion
Aus zyklischen Voltammetrieuntersuchungen im Potentialbereich von -800 mV bis +500 mV (Vorschubgeschwindigkeit 100 mV/s) in einem 2,0:1-Elektrolyten (nicht gezeigt) kann eine Coulomb-Effizienz von > 98 % berechnet werden, was die Reversibilität der Abscheidung und Auflösung von Aluminium belegt. Potentiodynamische Polarisationsexperimente zeigen, dass die Stromdichte bei +150 mV stark abnimmt und ein plateauartiger Bereich für einen 2,0:1-Elektrolyten folgt (Abb. 2(a), rote Kurve). Dieser Kurvenverlauf ist charakteristisch für die Passivierung der Oberfläche.

Abb. 2: Potentiodynamische Polarisationskurven einer Aluminiumelektrode in einem 1,5:1 (schwarz) bzw. 2,0:1 (rot) Elektrolyten (Vorschubgeschwindigkeit 0,1 mV/s) (a) und zyklisches Voltammogramm und Änderung von Frequenz, Dämpfung und Masse aus EQCM-Messungen (Vorschubgeschwindigkeit 1 mV/s) in einem 2,0:1-Elektrolyten (b) (die Interpretation der Farbverweise in der Abbildungslegende kann der Webversion dieses Artikels entnommen werden)
In einem 1,5:1-Elektrolyten (Abb. 2(a), schwarze Kurve), in dem die Gleichgewichtskonzentration von AlIII deutlich niedriger ist als in einem 2,0:1-Elektrolyten – was zu einer besseren Löslichkeit für weitere Aluminiumionen führt – erfolgt dagegen keine anodische Passivierung.
Mit zunehmender Vorschubgeschwindigkeit verschiebt sich das Passivierungspotential in anodische Richtung und die Spitzenstromdichte steigt (Abb. 2(a) und (b)). Die Abscheidung von Aluminium setzt bei –80 mV ein (Abb. 2(b)). Die abgeschiedene Masse nimmt im Potentialbereich von –80 mV bis –800 mV kontinuierlich zu. Die durchschnittliche Steigung der Kurve von Masse m gegen Ladung Q beträgt (84,3 ± 0,1) μg/C, was nahe am theoretischen Wert von 93,2 μg/C für reines Aluminium liegt. Der niedrigere Wert resultiert wahrscheinlich aus dem Einbau von leichteren Elementen (z. B. Kohlenstoff aus [EMIm]+). Die genaue Masse-Ladungs-Bilanz der Zersetzung von [EMIm]+ ist nicht bekannt [20, 21]. Eine Aussage über die Menge der eingebauten Verunreinigungen kann daher nicht getroffen werden. Die Dämpfung des Quarzes nimmt zu, ist aber deutlich geringer als die Frequenzabnahme (Abb. 2(b)). Daher ist Gleichung <2> gültig.
Eine mögliche Erklärung für diese leichte Erhöhung der Dämpfung ist, dass die Rauheit der Abscheidung höher ist als die der Goldschicht darunter. Dadurch steigt die Energiedissipation und die Dämpfung nimmt zu [15]. Kurz vor Erreichen des Umkehrpotentials von –800 mV oszilliert die Stromdichte und die Dämpfung des Quarzkristalls erreicht beim anodischen Scan bei etwa –700 mV ihr Maximum. Die Verringerung der Aluminiumionenkonzentration führt zu einer Abnahme der Lewis-Azidität und einem leichten Anstieg des Schmelzpunkts (Abb. 1), woraus eine höhere Elektrolytviskosität und damit eine höhere Dämpfung resultiert. Durch Verschieben des Potentials in anodische Richtung nimmt die Masse weiter zu. Die durchschnittliche Steigung des Verhältnisses aus Masse pro Ladung (Dm vs. DQ) liegt in diesem Bereich bei (77,0 ± 0,1) μg/C. Dies könnte auf eine elektrochemische Zersetzung von [EMIm]+ und den Einbau der Zersetzungsprodukte von [EMIm]+ hindeuten, die bei höheren kathodischen Potentialen stattfindet [20, 21].
Die Dämpfungsänderung nimmt bei zunehmend anodischer Polarisation ab. Im Vergleich zum Scan in kathodischer Richtung bleibt die Dämpfung höher, was auf die oben erwähnte Verarmung des Elektrolyten zurückzuführen ist. Liu et al. [20] berichteten von einer kornfeinenden Wirkung der Zersetzungsprodukte von [EMIm]+. Daher hat die Abscheidung eine glattere Morphologie. Rauheitseffekte [15] sollten dann vernachlässigbar sein, was zu einer abnehmenden Dämpfungsänderung bei zunehmend anodischer Polarisation führt, insbesondere unmittelbar nach dem Umkehrpotential von –800 mV. Eine anodische Spitzenstromdichte von 8,5 mA/cm2 wird bei 430 mV erreicht, was mit den potentiodynamischen Polarisationsmessungen übereinstimmt (Abb. 2(a)).
Bei anodischen Potentialen von mehr als 430 mV fällt die Stromdichte steil auf 35 µA/cm2 ab, was auf eine Passivierung hindeutet. Die EQCM-Daten zeigen eine starke Frequenzabnahme und Dämpfungszunahme, bevor die anodische Stromdichte abfällt (Abb. 2(b)). Für diese Erscheinung kann Gleichung <2> nur bedingt genutzt werden, da sie streng genommen nur dann gilt, wenn die Dämpfungsänderung kleiner als die Frequenzänderung ist. Da in diesem Potentialbereich kein kathodischer Strom fließt, kann eine elektrochemische Abscheidung jeglichen Materials ausgeschlossen werden. Chlorentwicklung würde einen verrauschten Stromdichtetransienten verursachen, der nicht beobachtet wurde und daher ebenfalls ausgeschlossen werden kann. Die Zunahme der Dämpfung weist jedoch auf eine Erhöhung des Produkts von Viskosität und Dichte des Elektrolyten hin (Gl. <3>).
Der Schmelzpunkt des Elektrolyten steigt stark an, wenn die molare Konzentration von Aluminiumchlorid (AlCl3) 67 Mol-% überschreitet (Abb. 1, Molverhältnis 2,0:1). Während der Schmelzpunkt eines 2,0:1-Elektrolyten bei etwa –90 °C liegt, liegt der Schmelzpunkt eines 2,5:1-Elektrolyten bei über 100 °C. Folglich könnte die steigende Zellspannung bei hohen anodischen Stromdichten ihre Ursache in einer lokalen Verfestigung des Elektrolyten aufgrund steigender Aluminiumkonzentration an der Anodenoberfläche haben. Vermutlich fällt der sich verfestigende Elektrolyt als schlecht leitender Feststoff aus und blockiert die aktive Elektrodenoberfläche. Folglich steigen die lokale Stromdichte an der aktiven Anodenoberfläche und das zum Aufrechterhalten der Ladungsübertragung erforderliche Potential. Dies führt zur Passivierung der gesamten Anodenoberfläche und steht im Einklang mit der abnehmenden Frequenz und zunehmenden Dämpfung der EQCM im anodischen Bereich des zyklischen Voltammogramms (Abb. 2(b)).
Außerdem erklärt dies die Verschiebung des Passivierungspotentials mit steigender Vorschubgeschwindigkeit. Während des kathodischen Scans wird der Elektrolyt an der Elektrodenoberfläche Lewis-basischer. Während des Scans in anodische Richtung diffundieren immer noch Aluminiumionen aus dem Elektrolytinneren zur Elektrodenoberfläche, wenn die anodische Auflösung von Aluminium einsetzt. Der lokal stark Lewis-basische Elektrolyt ist in der Lage, mehr Aluminium zu lösen, bevor die kritische Konzentration überschritten wird und eine lokale Verfestigung auftritt. Dieser Effekt ist umso stärker, je höher die Geschwindigkeit der Potenzialänderung ist, was zu einer Verschiebung des Passivierungspotentials zu anodischeren Werten mit steigender Vorschubgeschwindigkeit führt.
Wenn der Ursprung des beschriebenen Passivierungsphänomens die Verfestigung des Elektrolyten aufgrund erhöhter Aluminiumkonzentration vor der Anode ist, wird es hauptsächlich durch langsame Diffusion verursacht. Folglich sind stromkontrollierte Sprungexperimente geeignet, diese Theorie zu stützen. Der Diffusionskoeffizient der Al2Cl7−-Ionen wurde zu (7,1 ± 0,3)·10−11 m2/s bestimmt, ermittelt aus Untersuchungen unter Anwendung von strom- und potentialkontrollierten Sprungexperimenten (nicht gezeigt). Aus dem Zustandsdiagramm von AlCl und [EMIm]Cl (Abb. 1) lässt sich die kritische Aluminiumkonzentration ableiten, bei welcher der Elektrolyt bei gegebener Temperatur erstarrt. Für diese Berechnung wurde die Abszisse in Abbildung 1 vom Molenbruch x(AlCl3) in die molare Konzentration c(Al) unter Anwendung der Gleichungen <4> und <5> umgerechnet:
 <4>
<4>
 <5>
<5>
Der kritische Stoffmengenanteil, xcrit(AlCl3), wurde durch Interpolation der jeweiligen Punkte aus Abbildung 1 berechnet, deren Verbindungsgerade eine Temperatur von 27,5 °C oberhalb der Aluminiumkonzentration eines 2,0:1-Elektrolyten schneiden. Diese Konzentration xcrit(AlCl3) = 68,85 mol% entspricht ccrit = 7,24 mol/L.
Abbildung 3 zeigt die theoretischen Konzentrationsprofile eines 2,0:1-Elektrolyten vor einer Aluminiumanode bei unterschiedlichen Stromdichten und Zeiten. Die Darstellung basiert auf der Sand-Gleichung, die den Konzentrationsverlauf vor einer Elektrode beschreibt [10] (Gl. <6>).

Abb. 3: Konzentrationsprofil c(x,t) eines 2,0:1-Elektrolyten in der Nähe einer Aluminiumelektrode bei angelegten anodischen Stromdichten j von 5 mA/cm2 (Punktlinien), 10 mA/cm2 (gestrichelte Linien) und 20 mA/cm2 (durchgezogene Linien) in Abhängigkeit von der Auflösungszeit t von 30 s (schwarz), 60 s (rot) und 120 s (blau), basierend auf Gl. <6> [10] und unter Verwendung eines Diffusionskoeffizienten von (7,1 ± 0,3)·10−11 m2/s; die durchgezogenen horizontalen Linien repräsentieren ccrit (obere Linie) bzw. c* (untere Linie) für 27,5 °C (die Interpretation der Farbverweise in der Abbildungslegende kann der Webversion dieses Artikels entnommen werden)
Für Gleichung <6> werden die Parameter Konzentration c, Abstand von der Elektrode x, die Zeit nach dem Stromsprung t, die Gleichgewichtskonzentration des Elektrolyten c*, die angelegte Stromdichte j, die Zahl der übertragenen Elektronen z, die Faraday-Konstante F und der Diffusionskoeffizient D herangezogen. Die Konzentration von Aluminiumionen in der Nähe der Elektrode steigt schnell an, wenn eine konstant anodische Stromdichte angelegt wird. Bei einer niedrigen Stromdichte (z. B. 5 mA/cm2) wird die kritische Konzentration ccrit innerhalb eines Zeitfensters von einigen Minuten (2 min für 5 mA/cm2) nicht erreicht. Bei höheren Stromdichten (z. B. 10 mA/cm2 oder 20 mA/cm2) überschreitet die Konzentration den Grenzwert in weniger als einer Minute und die natürliche Konvektion hat keinen nennenswerten Einfluss auf das Konzentrationsprofil. Eine verfeinerte Theorie müsste einen verstärkten Massentransport aufgrund natürlicher Konvektion berücksichtigen, der das Konzentrationsprofil beeinflusst.
 <6>
<6>
Die kritische Transitionszeit tcrit kann als die erforderliche Zeit zum Erreichen der kritischen Konzentration ccrit an der Elektrodenoberfläche (x=0) definiert werden. Sie kann nach Gleichung <7>, basierend auf Gleichung <6>, bestimmt werden:
 <7>
<7>
Abbildung 4(a) zeigt die berechneten kritischen Transitionszeiten in Abhängigkeit von der angelegten Stromdichte und den gemessenen Transitionszeiten eines 2,0:1-Elektrolyten. Trotz einer geringfügigen Abweichung von den theoretischen Kurven stimmen die gemessenen Werte von tcrit gut mit dem diskutierten Modell überein. Über den gesamten Bereich der Stromdichten liegen die gemessenen Werte zwischen den theoretischen für 2,0:1- und 1,9:1-Elektrolyte. Etwas höhere kritische Transitionszeiten könnten das Ergebnis eines leicht verzögerten Beginns der Nukleation der erstarrenden Schmelze (Unterkühlung) sein, wie auch aus den EQCM-Messungen abgeleitet werden kann. Diese zeigen, dass die Dämpfung zunimmt, bevor die Frequenz abnimmt, begleitet von der Abnahme der Stromdichte (Abb. 2(b)).

Abb. 4: Kritische Transitionszeit tcrit (a) und inverse Quadratwurzel der kritischen Transitionszeit gegen die Stromdichte j (b) für 2,0:1 (schwarz), 1,9:1(rot), 1,8:1 (blau), 1,7:1 (violett), 1,6:1 (grün) und 1,5:1 (dunkelblau)-Elektrolyte. Die experimentellen kritischen Transitionszeiten (schwarze Punkte), die lineare Anpassung für tcrit < 1 min (rote gestrichelte Linie) und die Trendlinie (rote gepunktete Linie) sind ebenfalls gezeigt (die Interpretation der Farbverweise in der Abbildungslegende kann der Webversion dieses Artikels entnommen werden)
Außerdem ist zu berücksichtigen, dass Gleichung <6> nur für semi-infinite Diffusion gilt. Die theoretischen Werte in Abbildung 4 basieren auf der Regression der Dichte für verschiedene Elektrolytzusammensetzungen und den Diffusionskoeffizienten aus Untersuchungen mittels strom- und potentialkontrollierter Sprungexperimente (nicht gezeigt). Zudem beträgt der Massenunterschied des benötigten AlCl3 bei der Herstellung von 2,0:1-Elektrolyten und 1,9:1-Elektrolyten lediglich 2,0 bis 2,5 %.
Eine Abweichung zwischen theoretischen und experimentellen Daten beruht daher auf Fehlern in der Masse (Genauigkeit der verwendeten Waage betrug 1 mg) für die Herstellung der Schmelze. Auch die Interpolation der Daten für den Schmelzpunkt bei der Bewertung der kritischen Konzentration kann zu Abweichungen führen. Es konnten nur zwei Punkte des Phasendiagramms (Abb. 1) berücksichtigt werden. Der Unterschied in der Aluminiumkonzentration zwischen diesen beträgt etwa 1,6 mol-%. Daher ist der notwendige Aufwand zur Herstellung von Elektrolyten unterschiedlicher Zusammensetzung für diese beiden ziemlich hoch. Die Ergebnisse liegen jedoch in einem sinnvollen Bereich, wenn die durch die genannten Faktoren verursachten Fehler berücksichtigt werden.
Das Diagramm der Stromdichte gegen die inverse Quadratwurzel der kritischen Transitionszeit zeigt ein lineares Verhalten und schneidet den Koordinatenursprung (Abb. 4(b)), was auf eine Diffusionskontrolle hindeutet. Folglich wird die Passivierung der Anode durch langsame Diffusion von Aluminiumionen von der Anode in den Elektrolyten, lokale Verfestigung des Elektrolyten aufgrund der erhöhten Aluminiumkonzentration und dessen Niederschlag auf der Anode verursacht. Vergleichbare Effekte wurden für Hochtemperatursalzschmelzen berichtet [25, 26]. Da die Schmelztemperatur in einem kleinen Konzentrationsbereich bis etwa 130 °C stark ansteigt (Abb. 1), verbessert eine Temperaturerhöhung die anodische Auflösung von Aluminium in den Elektrolyten nur geringfügig. Selbst bei erhöhten Temperaturen wird die Auflösung von Aluminium in einem 2,0:1-Elektrolyten durch die Fähigkeit des Elektrolyten begrenzt, Aluminium aufzulösen, bevor der lokale Erstarrungspunkt überschritten wird. Darüber hinaus ist auch die thermische Zersetzung von [EMIm]+ ein
limitierender Faktor.
Die Löslichkeit verschiedener Metallsalze in dieser Art von ionischen Flüssigkeiten nimmt mit der Lewis-Azidität des Elektrolyten zu [1], was ein wichtiger Aspekt für die Abscheidung von Aluminiumlegierungen ist. Folglich muss ein Kompromiss zwischen Lewis-Azidität und der Tendenz des Elektrolyten zur anodischen Passivierung gefunden werden. Darüber hinaus könnte die Zugabe von anderen Metallsalzen als Aluminiumchlorid zu dieser Art von Elektrolyten auch eine Änderung des Schmelzpunkts bewirken [27, 28]. Die Auswirkung dessen muss für jeden einzelnen Elektrolyten individuell abgeschätzt werden.
Für kleine Stromdichten weichen die experimentell bestimmten kritischen Transitionszeiten von den theoretischen Werten ab, was durch das Einsetzen natürlicher Konvektion erklärt werden kann. Für das hier untersuchte System kann eine ungefähre kritische anodische Stromdichte von 8,5 mA/cm2 bei 27,5 ± 2,5 °C für einen 2,0:1-Elektrolyten abgeschätzt werden (Abb. 4(b)). Dieser Wert stellt die maximal anwendbare Stromdichte dar, um Transportbeschränkungen und damit die Passivierung der Anode für stationäre Abscheidungsbedingungen zu vermeiden. Dieser Wert wird auch in der potentiodynamischen Polarisationskurve eines 2,0:1-Elektrolyten (Abb. 2(a)) und im EQCM-gekoppelten zyklischen Voltammogramm (Abb. 2(b)) bestätigt.
4 Schlussfolgerungen
Während in einem 2,0:1-AlCl3-[EMIm]Cl-Elektrolyten eine anodische Passivierung auftritt, wird in einem 1,5:1-Elektrolyten keine Passivierung beobachtet. Die EQCM-Daten zeigen eine Frequenzabnahme und eine Dämpfungszunahme unmittelbar bevor die Stromdichte aufgrund der Passivierung abfällt, was auf eine Verfestigung und Ausfällung des Elektrolyten an der Elektrodenoberfläche hindeutet.
Stromkontrollierte Sprungexperimente zeigten, dass der Massentransport für die anodische Auflösung von Aluminium bei hohen Stromdichten limitierend wirkt. Eine hohe Auflösungsgeschwindigkeit von Aluminium bewirkt eine starke Erhöhung des Schmelzpunktes des Elektrolyten, der sich dann verfestigt und auf der Elektrode ausfällt, was gemäß den EQCM-Messungen eine Passivierung durch die Bildung eines schlecht leitfähigen Films verursacht. Bei Elektrolyten mit hoher Aluminiumkonzentration wird für Langzeitabscheidungsprozesse intensives Rühren oder ein hohes Verhältnis von Anoden- zu Kathodenfläche empfohlen, um eine anodische Passivierung zu verhindern.
Die Ergebnisse sind auf andere Elektrolyte übertragbar, die ein eutektisches Verhalten verbunden mit einer langsamen Diffusion und einer deutlichen Viskositätsänderung in Abhängigkeit von der Elektrolytzusammensetzung zeigen.
Kontakt:
Dr.-Ing. René Böttcher, TU Ilmenau
E-Mail: rene.boettcher@tu-ilmenau.de
Danksagungen
Die Autoren bedanken sich für die finanzielle Unterstützung des Bundesministeriums für Wirtschaft und Energie im Rahmen des Projekts NiCO (Kennzeichen 20W1523H) und die Unterstützung der Artikelbearbeitungsgebühr durch die Deutsche Forschungsgemeinschaft (DFG) und dem Open-Access-Publikationsfonds der Technischen Universität Ilmenau. Dank gilt auch Mohammadshahabaldin Najafi für seine Unterstützung bei den elektrochemischen Messungen.
Literatur
[1] F. Endres, A. Abbott, D. Mac Farlane: Electrodeposition From Ionic Liquids, 2. Auflage, Wiley-VCH, Weinheim, 2017
[2] G. R. Stafford, C. L. Hussey: Electrodeposition of transition metal-aluminum alloys from chloroaluminate molten salts; in: R. C. Alkire, D. M. Kolb (Eds.), Adv. Electrochem. Sci. Eng. 7, Wiley-VCH, Weinheim, 2002, S. 275–348
[3] Q. X. Liu, S. Z. El Abedin, F. Endres: Electroplating of mild steel by aluminium in a first generation ionic liquid: a green alternative to commercial Al-plating in organic solvents; Surf. Coat. Tech. 201 (2006), S. 1352–1356
[4] L. Simanavičius, A. Staknas, A. Šarkis: Codeposition of aluminum with some metals from AlBr3-dimethylethylphenylammonium bromide solutions containing acetylacetonate of selected metal; Electrochim. Acta 46 (2000), S. 499–507
[5] A. Ispas, E. Wolff, A. Bund: An electrochemical quartz crystal microbalance study on electrodeposition of aluminium and aluminium-manganese alloys; J. Electrochem. Soc. 164 (2017), S. H5263–H5270
[6] T. Jiang, M. C. Brym, G. Dubé, A. Lasia, G. M. Brisard: Electrodeposition of aluminum from ionic liquids: Part I - electrodeposition and surface morphology of aluminum from aluminum chloride (AlCl3)-1-ethyl-3-methylimidazolium chloride ([EMIm] Cl) ionic liquids; Surf. Coat. Tech. 201 (2006), S. 1–9
[7] A. Bakkar, V. Neubert: Electrodeposition and corrosion characterisation of micro- and nano-crystalline aluminium from AlCl3/1-ethyl-3-methylimidazolium chloride ionic liquid; Electrochim. Acta 103 (2013), S. 211–218
[8] H. M. A. Abood, N. L. Dawood: Morphology of electrodeposited aluminium metal from aluminium chloride-urea room temperature ionic liquid (RTIL) at variable parameters; Int. J. Sci. Res. (2015), S. 753–760
[9] C. Wang, A. Creuziger, G. Stafford, C. L. Hussey: Anodic dissolution of aluminum in the aluminum chloride-1-ethyl-3-methylimidazolium chloride ionic liquid; J. Electrochem. Soc. 163 (2016), S. H1186–H1194
[10] H. J. S. Sand III: On the concentration at the electrodes in a solution, with special reference to the liberation of hydrogen by electrolysis of a mixture of copper sulphate and sulphuric acid; Lond. Edinb. Dublin Philos., Mag. J. Sci. 1 (1901), S. 45–79
[11] F. G. Cottrell: Der Reststrom bei galvanischer Polarisation, betrachtet als ein Diffusionsproblem; J. Phys. Chem. 42U (1903), S. 386–431
[12] V. A. Isaev, Y. P. Zaykov, O. V. Grishenkova, A. V. Kosov, O. L. Semerikova: Analysis of potentiostatic current transients for multiple nucleation with diffusion and kinetic controlled growth; J. Electrochem. Soc. 166 (2019), S. D851–D856
[13] W. Lorenz: Oszillographische Überspannungsmessungen. I; Ber. Bunsenges, Phys. Chem. 58 (1954), S. 912–918
[14] V. M. Mecea: From quartz crystal microbalance to fundamental principles of mass measurements; Anal. Lett. 38 (2005), S. 753–767
[15] E. M. Moustafa, S. Z. El Abedin, A. Shkurankov, E. Zschippang, A. Y. Saad, A. Bund, F. Endres; Electrodeposition of Al in 1-butyl-1-methylpyrrolidinium
bis(tri-fluoromethylsulfonyl)amide and 1-ethyl-3-
methylimidazolium bis(tri-fluoromethylsulfonyl)amide ionic liquids; J. Phys. Chem. B 111 (2007), S. 4693–4704
[16] K. K. Kanazawa, J. G. Gordon: The oscillation frequency of a quartz resonator in contact with liquid; Anal. Chim. Acta 175 (1985), S. 99–105
[17] A. Ispas, A. Bund, F. Endres: Application of the electrochemical quartz crystal microbalance for the investigation of metal depositions from ionic liquids; ECS Trans. 16 (2009), S. 411–420
[18] M. Stich, N. Pandey, A. Bund: Drying and moisture resorption behaviour of various electrode materials and separators for lithium-ion batteries; J. Power Sources 364 (2017), S. 84–91
[19] A. Ispas, B. Adolphi, A. Bund, F. Endres: On the electrodeposition of tantalum from three different ionic liquids with the bis(trifluoromethyl sulfonyl) amide anion; Phys. Chem. Chem. Phys. 12 (2010), S. 1793–1803
[20] Q. X. Liu, S. Z. El Abedin, F. Endres: Electrodeposition of nanocrystalline aluminum; J. Electrochem. Soc. 155 (2008), S. D357–D362
[21] J. Tang, K. Azumi: Optimization of pulsed electrodeposition of aluminum from AlCl 3-1-ethyl-
3-methylimidazolium chloride ionic liquid; Electrochim. Acta 56 (2011), S. 1130–1137
[22] A. A. Fannin Jr, D. A. Floreani, L. A. King, J. S. Landers, B. J. Piersma, D. J. Stech, R. L. Vaughn, J. S. Wilkes, J. L. Williams: Properties of 1, 3-dialkylimidazolium chloride-aluminum chloride ionic liquids; J. Phys. Chem. 88 (1984), S. 2614–2621
[23] M. Zhang, R. Groves, R. M. Counce, J. S. Watson, T. A. Zawodzinski: Melting/freezing points of high concentrations of AlCl3 in a saturated chloroaluminate ionic liquid; J. Therm. Anal. Calorim. 124 (2016), S. 395–398
[24] H. A. Øye, M. Jagtoyen, T. Oksefjell, J. S. Wilkes: Vapour pressure and thermodynamics of the system 1-methyl-3-ethyl-imidazolium chloride – aluminium chloride; MSF 73–75 (1991), S. 183–190
[25] G. L. Holleck, J. Giner: The aluminum electrode in AlCl3-alkali-halide melts; J. Electrochem. Soc. 119 (1972), S. 1161–1166
[26] D. Orbakh: Nonaqueous Electrochemistry, 1. Auflage, Marcel Dekker, New York, 1999
[27] W. R. Pitner: Electrodeposition of zinc from the Lewis acidic aluminum chloride-1-methyl-3-ethylimidazolium chloride room temperature molten salt; J. Electrochem. Soc. 144 (1997), S. 3095–3103
[28] S.-I. Hsiu, J.-F. Huang, I.-W. Sun, Ch.-H. Yuan, J. Shiea: Lewis acidity dependency of the electrochemical window of zinc chloride–1-ethyl-3-methylimidazolium chloride ionic liquids; Electrochim. Acta 47 (2002), S. 4367–4372
