
Für die Messung der Korrosionsbeständigkeit von Chrom-Nickel-Schichten mit Chrom aus einem trivalenten Chromelektrolyten eignen sich verschiedene Methodiken. Hierbei haben die Abscheidedauer sowie Elektrolytverunreinigung von Eisen und Nickel Einfluss auf die Korrosionsbeständigkeit der Schichten. Zur Messung der Korrosionsbeständigkeit wurden Dauerversuche im Salzsprühnebelschrank (SSN) durchgeführt und mit den Ergebnissen der elektrochemischen Korrosionsmessungen verglichen. Verwendet wurden lineare Voltammetrie, elektrochemische Impedanzspektroskopie sowie Chronopotentiometrie. Außerdem wurden Farbe, Glanz und Schichtdicke in Abhängigkeit von den verschiedenen Abscheideparametern charakterisiert. Die Untersuchungen zeigten, dass sich der Salzsprühnebelschrank am besten zur Charakterisierung der Korrosionsbeständigkeit eignet. Elektrochemische Korrosionsmessungen lieferten zwar mehr Informationen, wiesen aber in ihren Ergebnissen teilweise noch Unklarheiten auf. Darüber hinaus wurde festgestellt, dass Eisenverunreinigungen im Elektrolyten die Korrosion am stärksten fördern.
1 Einleitung
Diverse Metalle, wie beispielsweise Nickel, Chrom oder Aluminium haben die Eigenschaft, bei Kontakt mit Sauerstoff eine selbstausheilende Oxidschicht zu bilden. Diese Eigenschaft ermöglicht diverse Anwendungsmöglichkeiten, insbesondere im Korrosionsschutz. Da die Oxidschicht elektrochemisch edler als der metallische Grundwerkstoff ist, wirkt sie passivierend und hemmt somit die Oxidation des Grundwerkstoffes. Eine Chromoxidschicht löst sich im Vergleich zu metallischem Chrom erst bei einem höheren anodischen Potential auf und schützt somit das Substrat vor weiterer Auflösung. Neben der guten Korrosionsbeständigkeit haben Chromschichten eine hohe Härte, sind elektrisch leitfähig, thermisch beständig und metallisch glänzend. All diese Eigenschaften ermöglichen, neben der reinen Anwendung zum Korrosionsschutz, vielseitige Möglichkeiten zur Nutzung von Chromschichten, beispielweise in der Fahrzeugindustrie, Sanitärindustrie und im Maschinenbau. Chrombeschichtungen sowie Chrom-Nickelbeschichtungen sind somit von hoher technischer Relevanz in der modernen Welt [1].
Konventionell werden in vielen Prozessen hexavalente Chromverbindungen im Abscheidungselektrolyt verwendet; hier liegt das Chrom in sechsfach oxidierter Form vor. Diese Verbindungen sind problematisch, da sie im Menschen krebserregend, stark reproduktionstoxisch wirken und zusätzlich stark umweltschädlich sind. Aufgrund dessen wurde die Verwendung von hexavalenten Chromverbindungen, wie beispielsweise Chromtrioxid, Chromsäuren und deren Salze, im Zuge der Verordnung (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) stark eingeschränkt [2].
Hier können trivalente Chromverbindungen eine nicht toxische Alternative darstellen, also Elektrolyte, in denen das Chrom in entsprechender Verbindung dreifach oxidiert vorliegt. Jedoch bringt der Umstieg von hexavalenten auf trivalente Chromverbindungen viele Schwierigkeiten mit sich. So weisen mit trivalenten Chromverbindungen hergestellte Schichten eine geringere Korrosionsbeständigkeit und eine abweichende Farbe auf. Des Weiteren lassen sich mit hexavalenten Elektrolyten sehr viel höhere Stromausbeuten und Abscheideraten erzielen. Während bei chrom(VI)basierten Prozessen Stromausbeuten von 15 % üblich sind, liegen diese Werte bei Elektrolyten auf Basis von Chrom(III)verbindungen bei etwa 5 %. Dementsprechend ergibt sich auch eine 3- bis 4-fach geringere Abscheiderate [2, 3] bei Verwendung von trivalenten Chromelektrolyten. Zusätzlich enthalten Chrom(III)elektrolyte eine höhere Anzahl an Komponenten und reagieren deutlich empfindlicher auf Verunreinigungen [4]. Hinzu kommt, dass die Methodiken zur Korrosionsmessung speziell an Schichtsystemen aus Elektrolyten auf Basis von Chrom(III) noch nicht so ausführlich untersucht worden sind, wie an chrom(VI)basierten Schichten [1].
Konventionell gibt es zwei Ansätze für die Messung der Korrosionsbeständigkeit: beschleunigte Korrosionstests in einer aggressiven Atmosphäre oder elektrochemische Korrosionstests. Bei den beschleunigten Korrosionstests wird das zu untersuchende Bauteil korrosiven Bedingungen ausgesetzt und die Korrosionsprodukte werden in bestimmten Zeitabständen dokumentiert. Bei elektrochemischen Messungen wird das Bauteil in der Regel als Anode geschaltet und resultierende Potentiale oder Wechselstromwiderstände werden ausgewertet. Elektrochemische Methodiken haben den Vorteil, dass verschiedene Parameter wie Stromdichte, Potential und Widerstand, direkt gemessen werden können, während sich bei den beschleunigten Tests nur Korrosionsprodukte auswerten lassen.
Vor dem Hintergrund des Umstiegs von hexavalenten zu trivalenten Chromverbindungen sollen die Ergebnisse von verschiedenen Methodiken der elektrochemische Korrosionsmessung, mit den Ergebnissen aus dem Salzsprühnebelschrank verglichen werden; Ziel ist es, die Korrosionsmessung an mittels Chrom(III)elektrolyten hergestellten Schichten besser zu verstehen.
2 Stand der Technik
Der Begriff Korrosion beschreibt die Zerstörung eines Bauteils durch eine chemische Reaktion mit seiner Umgebung, beispielsweise durch Feuchtigkeit oder Salze.
Elektrochemisch gesehen ist ein Korrosionsangriff eine Oxidation durch ein in der Elektrolytlösung vorhandenes Oxidationsmittel. In der Praxis handelt es sich bei dem Oxidationsmittel meist um Sauerstoff aus der Atmosphäre, der in Wasser gelöst ist [5]. Andere häufig vorkommende Oxidationsmittel sind Schwefeldioxid in Industrieatmosphäre oder Peroxoverbindungen in Waschmitteln [5]. Das oxidierende Medium wird reduziert, während das Metall oxidiert wird. Diese Abläufe sind in Gleichung <1> und <2> am Beispiel einer sehr gängigen Korrosionserscheinung, der Bildung von Rost an Eisen, dargestellt:
Oxidation: Fe → Fe2+ + 2e– <1>
Reduktion: O2 + 4e– + 2H2O → 4OH– <2>
In Wasser befindet sich neben diversen anderen Ionen auch meist Chlor, das als Katalysator für die Sauerstoffkorrosion wirken kann, indem es die Passivschicht destabilisiert und in deren Folge häufig lokale, lochartige Korrosion auftritt. Somit sind Bauteile, die ständiger Feuchtigkeit ausgesetzt sind, beispielsweise Wasserhähne oder Rohre, prädestiniert für Korrosion und benötigen deshalb einen ausreichenden Korrosionsschutz [1].
Beim passiven Korrosionsschutz existiert eine physikalische oder chemische Barriere zwischen dem Werkstoff und seiner Umgebung. So verhindern beispielsweise Oxidschichten oder Lackierungen, dass Elektrolyt, Sauerstoff oder andere Ionen mit der Metalloberfläche in Kontakt treten. Beim aktiven Korrosionsschutz wird das zu schützende Metall so beeinflusst, dass es nicht mehr als Anode fungieren kann; die Korrosion wird gezielt auf ein anderes unedleres Material verlagert, eine sogenannte Opferanode. Beispiele für aktiven Korrosionsschutz sind das Feuerverzinken und das galvanische Verzinken auf Eisenwerkstoffen, bei dem die Zinkschicht als Opferanode fungiert und dadurch der Eisenwerkstoff zur Kathode (kathodischer Korrosionsschutz) wird.
Erste Grundlagen für moderne Korrosionsforschung setzt U. R. Evans gegen 1930; er führt Potentialmessungen zur Charakterisierung der Korrosionserscheinungen ein und beschreibt Korrosion als einen lokal gekoppelten elektrochemischen Prozess [6, 7].
Die Charakterisierung der Korrosionsbeständigkeit beschreibt die zugrundeliegende Oxidationsreaktion. Bei der Anwendung von elektrochemischen Methoden können Oxidationsreaktionen zu jedem Zeitpunkt betrachtet werden; zudem lassen sich Korrosionsgeschwindigkeit und Korrosionsrate ermitteln. Eine gängige Methodik ist die Voltammetrie. Bei der Voltammetrie handelt es sich um die Aufnahme einer Strom-Spannungskurve. Ein Potential wird mit bestimmter Scangeschwindigkeit über einen definierten Potentialbereich durch das Bauteil geleitet. Parallel dazu wird in Bezug zu einer Referenzelektrode die resultierende Stromdichte aufgezeichnet. So kann charakterisiert werden, bei welchen Potentialen elektrochemische Reaktionen auf der Bauteiloberfläche stattfinden. Über Voltammetriekurven können die Korrosionsstromdichte, das Korrosionspotential und der Symmetriefaktor einer Reaktion ermittelt werden. Ein hohes Korrosionspotential weist auf eine geringere thermodynamische Korrosionsneigung hin und eine hohe Korrosionsstromdichte steht für eine hohe Korrosionsgeschwindigkeit.
Bei der elektrochemischen Impedanzspektroskopie wird ein sinusförmiges Potential durch das Bauteil geleitet und der resultierende Strom gemessen. Die Frequenz des Potentials variiert dabei logarithmisch im Bereich von typischerweise 102 Hz bis zu 106 Hz [8]. Alternativ kann auch ein Strom angelegt und die resultierende Spannung gemessen werden (galvanostatische Arbeitsweise). Für jede Frequenz werden nun Potentiale und Ströme über Zeit gemessen, mittels Fourier-Transformation werden Amplituden von Strom und Potential sowie die Phasenverschiebung des Stroms bei jeder Frequenz ermittelt. Im Nyquistplot wird der komplexe Wechselstromwiderstand dargestellt. Über den Durchmesser der resultierenden Halbkreise (Abb. 1) lassen sich die Widerstände ermitteln. Der ohmsche Widerstand der Elektrolytlösung wird durch Rs beschrieben, und ist im Ersatzschaltbild (Abb. 1) in Reihe geschaltet, da der Strom durch den Elektrolyt geleitet wird. An der Grenzfläche Elektrode/Elektrolyt bildet sich eine frequenzabhängige Doppelschichtkapazität (Cdl) aus. Der anliegende Strom wird anteilig zur Aufladung dieser Kapazität oder für den Ladungstransfer genutzt. Der Widerstand für den Ladungstransfer der elektrochemischen Reaktion wird mit Rct beschrieben. Je größer der Ladungstransferwiderstand ist, desto höher ist die Korrosionsbeständigkeit des untersuchten Bauteils.

Abb. 1: Nyquistplot schematisch (inks) und Ersatzschaltbild (rechts oben) [8] mit den Größen Elektrolytwiderstand (Rs), Ladungstransferwiderstand (Rct) und Doppelschichtkapazität (Cdl)
Eine weitere Methode ist die sogenannte Chronopotentiometrie (oder auch STEP-Test, STEP = Simultaneous Thickness and Electrochemical Potential). Hier wird die zu untersuchende Schicht als Anode geschaltet, sodass ein konstanter Strom durch das Bauteil fließt, während das resultierende Potential über die Zeit in Bezug zu einer Referenzelektrode gemessen wird. Da sich verschiedene Schichten bei verschiedenen Potentialen auflösen, beschreibt ein Potentialplateau den Prozess der Auflösung einer Schicht. Über die Zeit, die der Auflösevorgang gedauert hat, lässt sich über das Faradaysche Gesetz die Schichtdicke berechnen [9].
Der Salzsprühnebeltest nach DIN EN ISO 9227 stellt eine empirische Messmethode dar. In einer geschlossenen Kammer werden die Bauteile einem gleichmäßig fein verteilten Nebel aus einer Salzlösung ausgesetzt. Die Norm schlägt je nach untersuchtem Material drei verschiedene Prüflösungen vor (Tab. 1).

Für reproduzierbare Ergebnisse ist es wichtig, dass die Korrosion ausschließlich von der Salzatmosphäre ausgeht. Deshalb dürfen die Bauteile keinen Kontakt zueinander oder zu anderen Metallen oder dem Gestell haben. Des Weiteren ist darauf zu achten, dass die Bauteile so aufgehängt werden, dass das Kondensat gleichmäßig ablaufen kann und nicht auf andere Bauteile tropft [11].
3 Experimentelles
Für die durchgeführten Untersuchungen wurden Chrom-Nickel-Schichten einseitig auf Winkelblechen aus Kupfer abgeschieden. Bei der Abscheidung der Chromschicht wurden Abscheidedauer und somit Schichtdicke, sowie die Elektrolytverunreinigungen von Eisen und Nickel variiert (Tab. 2).

Jede Abscheidung wurde doppelt durchgeführt; eine Probe wurde im Salzsprühnebelschrank verwendet, eine für elektrochemische Messungen. Als Vorbehandlung wurde das Substrat vor jeder Abscheidung mit HSO Uni Entfetter bei 50 °C stromlos für 5 min entfettet und für 30 s bei Raumtemperatur in 10% -iger Schwefelsäure dekapiert. Um die Beschichtung einseitig aufzubringen, wurde eine Seite der Bleche mit galvanogeeignetem grünem Abdecklack abgedeckt.
Für die Nickelabscheidungen wurde der Elektrolyt Glanznickel Orion 3000+ der Kiesow Oberflächenchemie GmbH & Co KG verwendet. Es wurde eine Schichtdicke von 14 µm bei einer Stromdichte von 4,5 A/dm2, einer Temperatur von 57 °C und einem pH-Wert von pH 3,7 mit einer S-Nickel Gegenanode abgeschieden. Für die Chromabscheidungen wurde der TriMac Blue Elektrolyt der Mac-Dermid Enthone Industrial Solutions bei 7 A/dm2, 55 °C und einem pH-Wert von pH 3,4 unter Verwendung einer Metallmischoxid-Anode eingesetzt. Beide Abscheidungen fanden unter Elektrolytbewegung statt; in den Tabellen 3 und 4 sind die Zusammensetzungen der Elektrolyte aufgelistet.

Die Versuche im Salzsprühnebelschrank wurden bei einer Konzentration von 50 g/L NaCl, einer Temperatur von 35 °C und einem pH-Wert von pH 6,5 bis pH 7,2 durchgeführt. Die Proben wurden mittels einer Angelschnur am Gestell der Salzsprühkammer befestigt und nach 24 h, 48 h, 72 h, 96 h und 192 h entnommen und fotografiert. Verwendet wurde ein Salzsprühnebelschrank des Unternehmens Adwest Köhler, Typ:HK 400. Abbildung 2 zeigt den verwendete Salzsprühnebelschrank mit aufgehängten Proben.

Abb. 2: Salzsprühnebelschrank mit Winkelblechen
Für die Korrosionsmessungen wurde ein Potentiostat Modell SP-150 mit Booster von Bio-Logic SAS verwendet. Die Versuchsparameter wurden mit der Software EC-Lab (Version 11.52) eingestellt und gemessen. Für alle Messungen wurde ein konditionierter Silber/Silberchloridring als Referenz mit einem Potential von +0,2 V vs. NHE verwendet. Als Elektrolytlösung diente eine 0,5 mol/L Schwefelsäurelösung mit 1 mol/L NaCl.
Durchgeführt wurden die Versuche mit dem Couloscope V18 der Helmut Fischer GmbH und einer extern angeschlossenen Pumpe, die den Elektrolyt während aller Versuche bewegt hat. Die Voltammetrieversuche wurden in einem Potentialbereich von -0,3 V bis 0,5 V und mit einer Scangeschwindigkeit von 1 mV/s durchgeführt. Der Frequenzbereich für die Impedanzspektroskopie betrug 0,08 Hz – 100 000 Hz. Für die Steptestversuche wurden eine Auflösedauer von 4 min bei einem Strom von 10 mA gewählt. Jede Messung wurde zweimal durchgeführt. Eine Übersicht des verwendeten Versuchsaufbaus für die elektrochemischen Korrosionsmessungen ist in Abbildung 3 zu sehen.

Abb. 3: Versuchsaufbau Korrosionsmessungen
4 Darstellung und Diskussion der Ergebnisse
Für die Beurteilung der Versuche im Salzsprühnebelschrank wurde der Korrosionsangriff optisch bewertet. Eine Probe für den Umfang des Korrosionsangriffes nach 192 h im Salzsprühnebelschrank zeigt Abbildung 4. Die Ergebnisse der entsprechenden Korrosionsangriffe pro Abscheideparameter sind in Abbildung 5 zusammengestellt.

Abb. 4: Bewertung des Korrosionsangriffs von 1 bis 5 (5 = stärkster Korrosionsangriff)

Abb. 5: Korrosionsangriff bei Prüfung im Salzsprühnebelschrank nach 192 h Prüfdauer
Es ist zu erkennen, dass mit Eisenverunreinigungen hergestellt Proben den stärksten Korrosionsangriff aufweisen, wobei die Menge an Korrosionsprodukten mit steigender Eisenverunreinigungskonzentration ansteigt. Proben die in einem Elektrolyten mit Nickelverunreinigungen abgeschieden wurden, weisen einen moderaten Korrosionsangriff auf, während Proben, die ohne Verunreinigungen abgeschieden worden sind, am wenigsten korrodieren. Zwischen der Schichtdicke und dem Korrosionsangriff ist kein deutlicher Zusammenhang zu erkennen.
Nachfolgend werden die Ergebnisse der elektrochemischen Korrosionsuntersuchungen gezeigt, beginnend mit der linearen Voltammetrie. Die Messungen zeigten unabhängig von den Abscheideparametern Oxidationspeaks zwischen 0,4 V und 06 V vs. RHE bei Stromdichten von 10 A/dm2 bis 20 A/dm2 (Abb. 6).

Abb. 6: Lineare Voltammetrie / Stromdichte-Potentialkurven
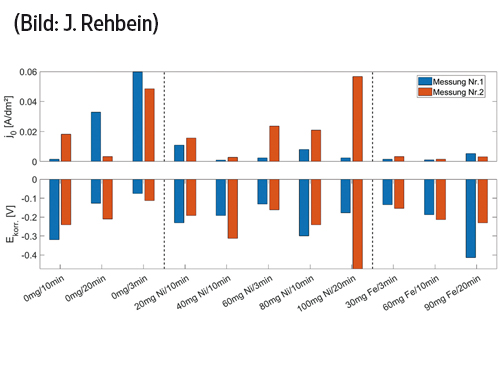
Für die weitere Auswertung wird die logarithmierte Stromdichte über die Überspannung dargestellt, anschließend wird der lineare Bereich dieser Kurve über die Bildung der ersten Ableitung bestimmt und extrapoliert. Über den y-Achsenabschnitt der extrapolierten Geraden kann die Korrosionsstromdichte und über ihre Steigung der Symmetriefaktor der Korrosion ermittelt werden. Der Schnittpunkt mit der x-Achse der logarithmierten Kurven ergibt den Wert für das Korrosionspotential. Der Ablauf ist in Abbildung 7 und 8 dargestellt. Die ermittelten Korrosionstromdichten und Potentiale zeigt die Abbildung 9. Der Symmetriefaktor lag unabhängig von den Abscheideparametern nahe 0,5.

Abb. 7: Logarithmus der Stromdichte (log(j)) als Funktion der Überspannung (h; blau) mit der ersten Ableitung(rot) und Ermittlung des linearen Bereichs

Abb. 8: Extrapolierte Kurve des Logarithmus der Stromdicht (log(j)) als Funktion der Überspannung (η)

Abb. 9: Korrosionsstromdichte j0 und Korrosionspotential Ekorr der untersuchten Schichten
Bei Proben hergestellt mit Eisen als Elektrolytverunreinigung ist zu erkennen, dass das Korrosionspotential mit zunehmender Schichtdicke und steigendem Gehalt an Elektrolytverunreinigungen zunehmend negativer wird. Je negativer das Korrosionspotential ist, desto geringer ist die Korrosionsbeständigkeit, womit die Ergebnisse der Versuche im Salzsprühnebelschrank bestätigt werden. Weitere deutliche Abhängigkeiten zwischen den unterschiedlichen Parametern sind nicht zu erkennen (Abb. 9).
Die Ergebnisse der Potentiometrie zeigen klare Potentialniveaus bei etwa 1,5 V (Abb. 10). Aus den Zeiten der Potentialniveaus lassen sich nach dem 2. Faradayschen Gesetz die entsprechenden Schichtdicken und anhand der Schichtdicken die Abscheideraten berechnen. Ermittelte Abscheideraten liegen zwischen 0,04 µm/min und 0,08 µm/min (Abb. 11). Tendenziell erhöhen sich die Abscheideraten mit steigender Konzentration an Elektrolytverunreinigungen.

Abb. 10: Ergebnisse der Potentiometrie bei einem Auflösestrom von 10 mA

Abb. 11: Abscheideraten bei den verschiedenen Abscheideparametern
Die Ergebnisse der Impedanzspektroskopie wiesen starke Störungen/Schwankungen auf, die wahrscheinlich durch einen unzureichenden Messaufbau resultierten. Die meisten Halbkreise waren kaum auswertbar (Abb. 12). Die ermittelten Ladungstransferwiderstände aus den auswertbaren Halbkreisen zeigten ebenfalls keine Abhängigkeit auf und sind hier nicht weiter aufgeführt.

Abb. 12: Nyquistplots mit einem vergrößerten Bereich (rotes Quadrat)
Außerdem wurde die Deckfähigkeit des Elektrolyten anhand einer grafischen Bildauswertung mittels Python untersucht. Hierfür wurden Bilder der hergestellten Proben auf einen definierten Bereich zugeschnitten und dann nach YUV, also hinsichtlich der Luminanz und Farbabweichung in blau-rot Richtung, ausgewertet. Folgend wurde nur noch der verfärbte Bereich im Knick der Bleche ermittelt. Die Bildauswertung in beiden Schritten ist in Abbildung 13 dargestellt. Die Deckfähigkeit scheint mit zunehmenden Nickelverunreinigungen zuzunehmen (Abb. 14).

Abb. 13: Grafische Auswertung mittels Python; Zuschnitt auf beschichtete Fläche (1) und Auswertung nach YUV einschließlich Ermittlung verfärbter Bereich (2)

Abb. 14: Korrelation Deckfähigkeit und Nickelverunreinigungen
Außerdem wurden die Farbe und der Glanz der Proben bei den verschiedenen Abscheideparametern gemessen. Für die Farbmessungen wurde der Lab*-Wert (L* = Schwarz/Weiß-Wert, a* = Rot/Grün-Wert, b* = Blau/Gelb-Wert) aufgenommen. Die Ergebnisse der Messungen sind in Abbildung 15 dargestellt, wobei die L*-Werte nicht aufgetragen sind, da sie für alle Proben zwischen 70 und 80 lagen.

Abb. 15: a* und b*-Werte für alle Proben nach Salzsprühnebelversuchen (links) sowie Farbkreis (rechts) [12]
Der a*-Wert (Rot/Grün-Wert) sinkt mit steigenden Mengen an Elektrolytverunreinigungen. Die Proben werden zunehmend gelblich mit steigender Konzentration an Eisen- und Nickelverunreinigungen im Elektrolyten.
Der Glanz der Proben im 20°-Winkel wurde vor und nach den Versuchen im Salzsprühnebelschrank ermittelt. Der 20°-Wert ist nach den Versuchen im Salzsprühnebelschrank sehr viel geringer (Abb. 16).

Abb. 16: Glanzmittelwerte für Winkel 20°, für alle im Salzsprühnebelschrank belasteten Proben
Der Glanz der Schichten nimmt durch die Korrosion ab. Der Wert der Abscheidung bei 10 min und ohne Elektrolytverunreinigung ist sehr gering, da hier eine Unterkonzentration an Glanzzusätzen im Nickelelektrolyten vorlag, die danach korrigiert wurde. Eine Abhängigkeit zwischen den Glanzwerten und den Abscheideparametern ist nicht zu erkennen.
5 Zusammenfassung
Die Ergebnisse der Korrosionsuntersuchungen im Salzsprühnebelschrank zeigen, dass Eisenverunreinigungen im einem trivalenten Chromelektrolyten die Korrosionsbeständigkeit der Chromschichten stark reduzieren. Da nach einer Prüfdauer von 192 h noch nicht alle Proben korrodiert sind, sollten für zukünftige Prüfungen längere Prüfzeiten verwendet werden. Mit den elektrochemischen Methodiken konnte dieses Ergebnis nicht direkt bestätigt werden. So zeigten mit Eisenverunreinigungen hergestellte Proben zwar das niedrigste Korrosionspotential, jedoch konnte keine Abhängigkeit zwischen den Abscheideparametern und der Korrosionsbeständigkeit in der Korrosionsstromdichte oder dem Ladungstransferwiderstand gefunden werden.
Generell wiesen die elektrochemischen Messmethodiken erhebliche Instabilitäten auf, was eine eindeutige Bewertung der Korrosionsmechanismen erschwert. Als mögliche Ursache könnten inhomogene Schichtzusammensetzungen, eine unregelmäßige Ausbildung der Passivschicht und ein unzureichender Messaufbau zugrundeliegen. Außerdem ist nicht auszuschließen, dass sich die elektrochemischen Eigenschaften der Chromschichten durch die Bildung von Metallmischoxiden verändern.
Über die Chronopotentiometrie (oder auch STEP-Test) konnten die Schichtdicken der meisten Proben anhand der Potentialplateaus abgelesen werden. Ein Vergleich mit RFA-Schichtdickenmessungen derselben Proben zeigte, dass die ermittelten Schichtdicken und Abscheideraten passend sind. Jedoch wurden vereinzelt auch nicht auswertbare Kurven gemessen, in denen kein deutliches Potentialniveau zu erkennen war.
Aus diesen Untersuchungsergebnissen lässt sich schließen, dass der Test mit Salzsprühnebelschrank ohne viel Vorarbeit zu betreiben ist und die am besten geeignete Methodik darstellt, um schnell einen Überblick über die Korrosionsbeständigkeit der abgeschiedenen Schichten zu erlangen. Für ein detailliertes Verständnis stellen Voltammetrie und elektrochemische Impedanzspektroskopie geeignete Methodiken dar. Jedoch müssen die Messparameter noch besser angepasst werden, um reproduzierbare und belastbare Ergebnisse zu erhalten.
6 Ausblick
Die erhaltene Ergebnisse bieten zahlreiche Ansätze für weiterführende Untersuchungen. So können elektrochemische Korrosionsmessungen mit alternativen Messelektrolyten, beispielsweise Phosphorsäure, durchgeführt werden, um elektrolytspezifische Effekte auf das Korrosionsverhalten zu identifizieren. Weiterführend könnten Untersuchungen an Proben mit zuvor entfernter Oxidschicht helfen, den Einfluss einer inhomogen ausgeprägten Oxidschicht auf das Korrosionsverhalten besser zu verstehen. Zu diesem Verständnisgewinn können auch Voltammetriemessungen bei veränderten Messparametern beitragen. Zusätzlich könnte eine Analyse der Zusammensetzung der Schichten, beispielsweise mittels GDOES, helfen, ein besseres Verständnis des Korrosionsverhalten zu erlangen und den Einbau der Elektrolytverunreinigungen in die Schicht zu spezifizieren.
Literatur
[1] J. Rehbein: Korrosionsmessungen an mittels Cr(III)-Elektrolyt hergestellten Chrom-Nickel-Schichten; Forschungsprojekt, Technische Universität Ilmenau, 2025
[2] M. Leimbach: Charakterisierung der elektrochemischen Abscheidung von Chrom aus Chrom(III)-Elektrolyten für dekorative Anwendungen; Dissertation, Technische Universität Ilmenau, 2022
[3] D. L. Snyder: Decorative chromium plating basics. Metal Finishing; Bd. 110, Nr. 2, S. 14–21, 2012
[4] Zentralverband Oberflächentechnik e. V.: Digitale Werkzeuge zur Verbesserung galvanischer Schichten am Beispiel chrom(III)-basierter Prozesse; ZVOreport, 2024
[5] H. Kaesche: Die Korrosion der Metalle; Springer Verlag, Berlin, 1990
[6] Institute of Corrosion: A Journey into the World of Corrosion Science; online verfügbar unter: https://www.icorr.org/world-corrosion-science/, Zugriff am 27.01.2025
[7] R. P. Frankenthal: A Brief History of Corrosion Science and Its Place in the Electrochemical Society; Basking Ridge, NJ, USA
[8] PINE Research Instrumentation: Electrochemical Impedance Spectroscopy (EIS) Basics; online verfügbar unter: https://pineresearch.com/shop/kb/theory/eis-theory/eis-basics, Zugriff am 27.01.2025
[9] Helmut Fischer GmbH: Coulometrisches Messverfahren; online verfügbar unter: https://www.helmutfischer.com/de/anwendungen/messverfahren/coulometrisches-messverfahren-und-steptest, Zugriff am 27.01.2025
[10] DIN EN ISO 9227: Korrosionsprüfungen in künstlichen Atmosphären – Salzsprühnebelprüfungen; Beuth Verlag, Berlin, 2017
[11] Kluthe GmbH: Was ist der Salzsprühnebeltest? online verfügbar unter: https://kluthe.com/magazin/was-ist-der-salzspruehnebeltest, Zugriff am 28.01.2025
[12] Sensor Instruments Entwicklungs- und Vertriebs GmbH: What is Color? online verfügbar unter: https://www.sensorinstruments.de/whatiswhat.php?subpage=11&language=en, Zugriff am 28.01.2025
Kontakt
Jonas Rehbein, TU Ilmenau, Fachgebiet für Elektrochemie und Galvanotechnik, Tel.: 03677693102,
E-Mail: jonas.rehbein@tu-ilmenau.de
